荷瘤小鼠口服舞茸中可溶性β-葡聚糖能诱导全身
日期:2019-07-31 / 人气:
声明:本文摘自International Journal of Cancer,即国际癌症杂志,是国际癌症控制联盟的官方杂志,每年24期。其内容主要是与癌症相关的实验室和临床研究,发表原创的研究论文,综述和报道。为方便广大读者阅读,我们对部分内容进行了中文翻译,译文仅供参考,如有不足之处,敬请指正。
【参考译文】
Oral administration of soluble β-glucans extracted from Grifola frondosa induces systemic antitumor immune response and decrease immunosuppression in tumor-bearing mice
荷瘤小鼠口服舞茸中提取的可溶性β-葡聚糖能诱导全身抗肿瘤免疫反应并减轻免疫抑制
舞茸蛋白多糖是一种从舞茸(一种东方食用菌)中提取的高纯度可溶性β葡聚糖。有研究表明腹腔注射舞茸蛋白多糖能通过增强宿主免疫系统从而抑制肿瘤生长。在本次试验中,我们证实在小鼠模型中口服舞茸蛋白多糖与腹腔注射一样具有显著抑制肿瘤生长的作用。口服舞茸蛋白多糖后,它并不是自由地转移到血液中,而是被抗原递呈细胞如巨噬细胞、树突细胞(DCs)等捕获并递呈到集合淋巴小结。这些捕获的舞茸蛋白多糖被输送到脾脏,从而激活全身的免疫系统。我们的试验显示舞茸蛋白多糖通过C型凝集素受体dectin-1路径诱导DC成熟,口服舞茸蛋白多糖的治疗反应与以下几点有关:(1)通过dectin-1介导使DCs(树突细胞)激活从而诱导全身肿瘤抗原特异性T细胞反应;(2)更多的已激活的T细胞渗透进入肿瘤;(3)减少因肿瘤引起免疫抑制的细胞数量如调节性T淋巴细胞、髓系来源抑制细胞。这些临床前的试验证明舞茸蛋白多糖对于癌症病人的治疗是一种有效的口服药物。
注:dectin-1为β-葡聚糖受体。
论文原文
International Journal of Cancer:108-120(2013)
Oral administration of soluble β-glucans extracted from Grifola frondosa induces systemic antitumor immune response and decreases immunosuppression in tumor-bearing mice
Yuki Masuda1, Hiroko Inoue1, Hiroya Ohta2, Ayumi Miyake2,
Morichika Konishi1,2 and Hiroaki Nanba1
1Department of Microbial Chemistry, Kobe Pharmaceutical University, 4-19-1, Motoyama-kitamachi, Higashinada-ku, Kobe, 658-8558, Japan
2Department of Genetic Biochemistry, Kyoto University Graduate School of Pharmaceutical Sciences, Sakyo, Kyoto, 606-8501, Japan
Maitake D (MD)-Fraction is a highly purified soluble β-glucan derived from Grifola frondosa (an oriental edible mushroom). Intraperitoneal (i.p.) injection of MD-Fraction has been reported to inhibit tumor growth via enhancement of the host immune system. In this study, we demonstrated that oral administration of MD-Fraction as well as i.p. injection significantly inhibited tumor growth in murine tumor models. After oral administration, MD-Fraction was not transferred to the blood in its free form but was captured by antigen-presenting cells such as macrophages and dendritic cells (DCs) present in the Peyer’s patch. The captured MD-Fraction was then transported to the spleen, thereby inducing the systemic immune response. Our study showed that MD-Fraction directly induced DC maturation via a C-type lectin receptor dectin-1 pathway. The therapeutic response of orally administered MD-Fraction was associated with (i) induced systemic tumor-antigen specific T cell response via dectin-1-dependent activation of DCs, (ii) increased infiltration of the activated T cells into the tumor and (iii) decreased number of tumor-caused immunosuppressive cells such as regulatory T cells and myeloid-derived suppressor cells. Our preclinical study suggests that MD-Fraction is a useful oral therapeutic agent in the management of patients with cancer.
Key words: β-glucan, Grifola frondosa, dectin-1, oral administration
Many approaches to tumor immunotherapy and vaccination have focused on enhancement of effector T cell responses by targeting of innate immune cells and the receptors that mediate their activation. The agonists for pattern-recognition receptors (PRRs), such as toll-like receptors (TLRs), nucleotide oligomerization domain and the dectin-1 receptors, are potent adjuvants for infectious disease and have been used as immunotherapeutic agents in cancer patients.1,2 β-Glucans are naturally occurring carbohydrates found in fungi, mushrooms, yeast, algae, lichens
and plants. Some natural medicines containing β-glucan have immunomodulatory effects and are used clinically for tumor immunotherapy in several countries.3,4 β-Glucans are homopolymers of D-glucose with β-1,3 linkages and varying number of β-1,6 linkages, polymer lengths and distribution. The recognition of β-glucans is thought to involve multiple interactions with PRRs, including dectin-1, complement receptor 3 (CR3), mannose receptor, scavenger receptors and lactosylceramide.5
Maitake D (MD)-Fraction, a soluble β-(1,3) (1,6)-glucan, highly purified from the maitake mushroom (Grifola frondosa), induces antitumor activity after intraperitoneal (i.p.) injection.6,7 Recently, MD-Fraction was shown to increase GM-CSF production in resident macrophages, resulting in proliferation and dectin-1-mediated tumor necrosis factor (TNF)-α production.8 MD-Fraction enhances granulopoiesis in granulocytopenic mice by oral administration and i.p. injection.9,10 A phase I/II trial using a polysaccharide extract from maitake mushrooms in the United States showed that oral administration of the extract affected the host immune system without dose-limiting toxicity.11 Oral administration of MD-Fraction may be a useful dietary supplement for healthy individuals to prevent cancer as well as an adjuvant for cancer therapy. However, because mammal digestive enzymes do not digest β-glucans, the mechanism by which MD-Fraction is absorbed in the gastrointestinal tract and how it functions to enhance the systemic immune response are unclear. The first effect of orally administered MD-Fraction is likely observed in the gut-associated lymphoid tissue (GALT). In general, M cells in Peyer’s patches (PPs) act as a portal for uptake of antigens from the intestinal lumen and subsequent transfer to the PPs. The molecular events of the PPs and mesenteric lymph nodes (MLNs) after oral administration of antigens are thought to induce the adaptive immune response.12
Although MD-Fraction can induce hematopoiesis and related cytokine induction after oral administration, there is little evidence that orally administered MD-Fraction can induce antitumor immunity. Antitumor immunity is initiated by dendritic cells (DCs) that capture tumor antigens from tumor cells and induce the CD4+ T helper (Th)-1 cell/CD8+ cytotoxic T cell (CTL) response.13 Both circulating and tumor-infiltrating tumor antigen-specific T cells are linked to superior antitumor efficacy in vivo.14 However, the induction of effective antitumor immune responses is hampered by the immunosuppressive nature of the tumor microenvironment, which is mediated by regulatory T (Treg) cells and myeloid-derived suppressor cells (MDSCs).15,16 The tumor microenvironment also inhibits DC maturation through the secretion of IL-10 and TGF-β, which causes antigen-specific anergy.17 Thus, successful immunotherapy likely requires both the increase of tumor-specific immunity and reversal of tumor-associated immune suppression.
In this study, we investigated how orally administered MD-Fraction interacts with the GALT and induces the systemic immune system. In addition, we investigated whether MD-Fraction influenced tumor microenvironment and evaluated the efficacy of oral MD-Fraction treatment in murine tumor models.
Material and Methods
Mice
Female BALB/c, BALB/c Nude and C3H/HeJ mice were purchased from CLEA Japan (Higashiyama, Japan), and 5-8-week-old mice were used in the study. Animal care and processing were approved by the animal care committee at the Kobe Pharmaceutical University.
Preparation of MD-Fraction
MD-Fraction was prepared from maitake fruiting bodies (Yukiguni Maitake, Niigata, Japan) as previously described.6,8 Briefly, MD-Fraction was isolated from boiling water extract of maitake fruiting bodies, followed by ethanol precipitation, alkaline extraction after acid precipitation and protein depletion using the Sevag method. The water-soluble fraction, MD-Fraction, showed a single peak with a MW of 1,200-2,000 kDa and consisted of 98% polysaccharide and 2% peptide. In addition, we tested for LPS contamination using the Endospecy ES-24S set (Seikagaku Biobusiness, Tokyo, Japan) and did not detect LPS contamination in the prepared MD-Fraction. A DTAF (Sigma-Aldrich)-labeled MD-Fraction (DTAF-MD-Fraction) was prepared as described previously.8
Tumor models
Colon-26 cells (1×105cells/mouse) and MM-46 cells (1×106 cells/mouse) were inoculated in BALB/c and C3H/HeJ mice, respectively, by subcutaneously (sc) injection. Tumor-inoculated mice were orally administered either phosphate-buffered saline (PBS) or MD-Fraction (20 mg/kg/day) once every 19 days after tumor inoculation, using an intragastric gavage needle. The tumor volume was calculated using the following formula: mm3= (longest diameter × shortest diameter2)/2.
Flow cytometry
Spleen, PP and MLN single-cell suspensions were prepared by filtration through 70 μm nylon strainers (BD Biosciences). Tumor tissues were minced and then digested in RPMI medium containing collagenase (70 units/mL; Wako Pure Chemical Industries, Osaka, Japan) for 45 min at 37℃. Tumor cells were filtered and washed twice with PBS and used as tumor cell suspensions. The cells were blocked with Fc receptor-blocking mAb and stained with mAb including CD4, CD8, CD49b, CD3, B220, CD11c, CD80, CD86, I-A/I-E, CD11b, Gr-1 (BD PharMingen, San Diego, CA) or F4/80 (AbD Serotec, Kidlington, UK) for 20 min at 4℃, then washed and analyzed using a flow cytometer (FACSCalibur; BD Biosciences). Intracellular staining of IFN-γ was performed using the Cytofix/Cytoperm kit (BD PharMingen). To evaluate CD4+Foxp3+ Tregs, cells were first stained with FITC-conjugated anti-CD4 and then permeabilized and stained using PE-conjugated anti-Foxp3 Abs (BD PharMingen).
Measurement of serum β-glucan
DTAF-MD-Fraction was orally administered into BALB/c mice, blood was collected from the tail vein of these mice at 0, 4, 12 and 24 hr after administration, and the serum was analyzed. DTAF-MD-Fraction in serum samples and DTAF-MD-Fraction standards (0.01-1,000 μg/mL) were measured at 485/520 nm. The Fungitec® G Test method, which can detect β-(1,3)-glucans from 3.9 to 500 pg/mL, was used as an alternative test according to the manufacturer’s instructions (Seikagaku Biobusiness).
Generation of bone marrow (BM)-derived DCs
BM-derived DCs were generated as described previously.18 More than 85% of the cells expressing CD11c were used. DCs (1 × 106cells/mL) were stimulated with MD-Fraction (400 μg/mL) for 24 hr. For blocking antibody assays, DCs were preincubated with blocking antibodies (5 μg/mL) for 1
h, followed by stimulation with MD-Fraction for 24 hr as recently described.8 The following blocking antibodies were used: anti-CD11b (M1/70; BioLegend, San Diego, CA) and anti-Dectin-1 (2A11; AbD Serotec).
Quantitation of cytokine production
IL-12p70 and IL-10 secretion was determined using ELISA kits according to the manufacturer’s protocols (IL-12p70; R&D Systems, Abingdon, UK, IL-10; PeproTech, Rocky Hill, NJ). IFN-γ levels were determined by a sandwich ELISA as previously described.18
Real-time PCR (RT-PCR) was used to examine mRNA expression levels of various cytokines. RNA was purified using RNeasy (Qiagen, Hilden, Germany) prior to first-strand cDNA synthesis using the ReverTra Ace qPCR RT kit (Toyobo, Osaka, Japan). RT-PCR using Thunderbird SYBR qPCR mix (Toyobo) was performed using primers for 18s, IFN-γ, IL-12p40, IL-4 and IL-10 as described previously.8 Other primes sequences are as follows: IL-17 forward primer 5’-TCCAGAAGGCCCTCAGACTA-3’, IL-17 reverse primer 5’-TGAGCTTCCCAGATCACAGA-3’; TGF-β forward primer 5’-GGAGAGCCCTGGATACCAAC-3’, TGF-β reverse primer 5’-CAACCCAGGTCCTTCCTAAA-3’; Arg 1 forward primer 5’-AGAGATTATCGGAGCGCCTT-3’, Arg 1 reverse primer 5’-TTTTTCCAGCAGACCAGCTT-3’; and inducible nitric oxide synthase (iNOS) forward primer 5’-TGAAGAAAACCCCTTGTGCT-3’, iNOS reverse primer 5’-TTCTGTGCTGTCCCAGTGAG-3’.
Analysis of tumor-specific immunity
Tumor-Ag specific T-cell proliferation was determined by a CFSE dilution assay.19 Briefly, CD4+ T cells were labeled with 5 μM CFSE (Invitrogen, Carlsbad, CA) for 15 min at 37℃. After washing, labeled cells (1 × 105 cells/well) were seeded in 96-well, U-bottom plates and were incubated with DCs (1 × 104 cells/well) for 72 hr. The preparation of tumor cell lysates and priming DCs were described previously.18 The CTL assay was performed as previously described.20
Statistical analysis
The presented data are expressed as the mean±standard error. One-way analysis of variance (ANOVA) with the Bonferroni post-test was used for analysis of multiple groups, and Student’s t-test was used to compare two groups. Tumor volumes of different groups were analyzed using a nonparametric 2-tailed test (Mann-Whitney test) for unpaired samples. p values<0.05 were considered statistically significant.
Results
Orally administered MD-Fraction inhibits tumor growth in murine cancer models
The antitumor activity of orally administered MD-Fraction was examined in two models of established murine tumors. MM46 mammary carcinoma or colon-26 adenocarcinoma cells were implanted in the right flank of C3H/He or Balb/c mice, and the tumor sizes were monitored. MD-Fraction was administered orally via a gastric gavage once daily, starting 1 day after the tumor inoculation. As shown in Figure 1, the tumor growth was retarded at doses of 20 mg/kg (680.9±40.9 mm3; p=0.36) and 80 mg/kg (534.2±50.2 mm3; p<0.05) of MD-Fraction-treated C3H/He mice of on day 15 after the inoculation with MM46 cells, compared with the PBS control
(830.0±91.7 mm3). On day 20 after the inoculation with colon-26 cells in Balb/c mice, the tumor volumes in mice receiving 5 mg/kg (643±106 mm3; p=0.01) and 20 mg/kg (412±38mm3; p<0.001) MD-Fraction were significantly smaller than those in mice receiving the PBS control (1,156±1,104 mm3).
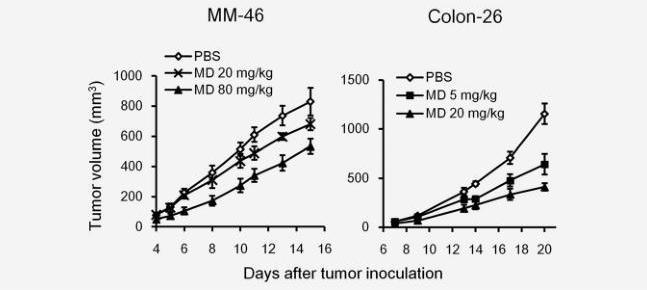
Figure 1. Orally administered MD-Fraction inhibits tumor growth. Mice were s.c. inoculated with MM-46 or colon-26 cells. MD-Fraction was orally administered to the mice 1 day after tumor inoculation, and the tumor growth was measured (n=3–6). The data presented are representative of three independent experiments.
DC and macrophages in the GALT capture the orally administered MD-fraction
We previously observed that DTAF-MD-Fraction was immediately detected in serum and peaked at 4 hr (33.1 μg/mL) after the i.p. injection (8 mg/kg). In addition, DTAF-MD-Fraction was phagocytosed by resident macrophages after the i.p. injection (unpublished observation). We next investigated the distribution of DTAF-MD-Fraction (20 mg/kg) after oral administration. Blood was collected at 0, 4, 12 and 24 hr after administration, and the serum was measured; however, DTAF-MD-Fraction was not detected. The minimum detectable dose of DTAF-MD-Fraction ranged from 0.05-0.1 μg/mL. To detect lower levels of MD-Fraction, we examined serum MD-Fraction by the Fungitec® G Test method, which can detect β-(1,3)-glucans at concentrations from 3.9 to 500 pg/mL. We confirmed that MD-Fraction was detectable at concentration>5 pg/mL
using this method. After oral administration of MD-Fraction (20 or 100 mg/kg), serum β-glucan concentrations were varied 8-13 pg/mL. No significant differences were observed between the initial β-glucan concentrations and the concentration at 4-24 hr. In addition, continuous administration of MD-Fraction (20 mg/kg) for 7 days did not change the serum β-glucan concentration. These results indicate that orally administered MD-Fraction is likely not transferred to the blood in its free form.
Orally administered MD-Fraction was not adsorbed directly into the portal vein; however, it is possible that MD-Fraction was captured by macrophages and DCs in the GALT. We therefore examined the percentage of DTAF-MD-Fraction+ cells in the PP and spleen by flow cytometry on 1, 3 and 7 days after the oral administration of DTAF-MD-Fraction (20 mg/kg/day). As shown in Figure 2a, DTAF-MD+ T cells (CD3+) and B cells (B220+) were not detected in the PP and spleen. In contrast, DTAF-MD+ macrophages (F4/80+) and DCs (CD11c+) were detected in the PP 1 day after the administration. In addition, 3 day after the administration, DTAF-MD-Fraction reached the macrophages and DCs in the spleen. These results suggest that the orally administered MD-Fraction is captured by macrophages and DCs in the GALT is then transferred to the systemic immune organs.
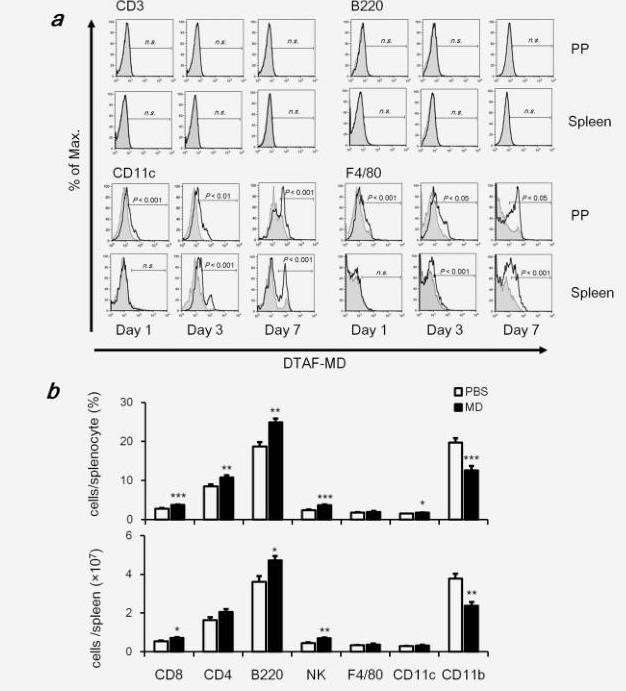
Figure 2. Orally administrated MD-Fraction is captured by DCs and macrophages in the GALT and then transferred to the systemic immune organs, affecting the systemic immune system. (a) Representative histograms of DTAF+ cells gated on CD11c, F4/80, CD3 or B220+ cells. The black line represents the DTAF fluorescence of cells from mice orally treated with MD-Fraction. The shaded gray line below represents the autofluorescence of cells from PBS control mice. Significances (p) indicate differences in the percentage of positive cells between the PBS control and MD-Fraction-treated mice. (b) On day 20 after colon-26 inoculation in BALB/c mice, splenocytes were stained with specific antibodies and analyzed by flow cytometry. Percentages and numbers of each cell in a whole-spleen cell suspension are shown. The data presented are representative of 2 independent experiments.
Orally administered MD-Fraction induces systemic immune responses
To examine whether orally administrated MD-Fraction influences the systemic immune response, BALB/c mice were orally administered 20 mg/kg MD-Fraction, and the spleen cells were analyzed on day 20 after the inoculation with colon-26 tumor cells. The total cell number in the spleen did not change by administration of MD-Fraction, but the percentage of T lymphocyte subsets (CD4+ and CD8+), B cells (B220+), NK cells (CD49b+CD3-) and DCs (CD11c+) in the spleen from MD-Fraction-treated mice were significantly increased compared with those in the PBS-treated mice (Fig. 2b). In addition, MD-Fraction significantly increased the cell number of splenic CD8+ T cells from 0.53 to 0.70 (×107, p=0.02), B cells from 3.6 to 4.7 (×107, p=0.02) and NK cells from 0.45 to 0.69 (×107, p<0.001) compared with PBS-treated mice. In contrast, MD-Fraction dramatically decreased the number of splenic myeloid cells (CD11b+) from 3.8 to 2.4 (×107, p<0.001). These results suggest that orally administered MD-Fraction modulates the systemic immune system.
Orally administered MD-Fraction induces CD4+ and CD8+ T cell responses against tumor cells
We next investigated mRNA levels of Th-associated cytokines in the PP, MLN, spleen and tumor by quantitative RT-PCR. As shown in Figure 3a, MD-Fraction significantly increased the expression of IL-12 in MLNs (1.6-fold, p=0.009), spleens (1.5-fold, p=0.02) and tumors (3.9-fold,
p=0.001) compared with the PBS group. In addition, MD-Fraction significantly increased the expression of IFN-γ in PPs (1.6-fold, p=0.004), MLNs (1.8-fold, p<0.001), spleens (1.5-fold, p=0.02) and tumor (2.5-fold, p=0.04). However, MD-Fraction did not alter the expression of IL-4 and IL-17 (Fig. 3a). These results show that orally administered MD-Fraction induces the systemic Th1 response, but not the Th2 and Th17 responses. To demonstrate that oral MD-Fraction induced antitumor immunity, we assessed whether IFN-γ-expressing CD4+ and CD8+ T cells in tumor-bearing mice increased due to oral administration of MD-Fraction on day 20 after tumor inoculation. Intracellular cytokine staining showed that mice administered MD-Fraction had more IFN-γ-expressing CD4+ in MLNs (p=0.004) and spleens (p<0.001) and IFN-γ-expressing CD8+ T cells in MLNs (p=0.004), spleens (p<0.001) and tumors (p<0.001) as compared with the PBS control mice (Figs. 3b and 3c). In addition, the CTL activity in splenocytes from MD-treated mice was significantly higher than in those from PBS-treated mice (Fig. 3d).
The number and type of leukocytes in the tumor infiltrate are related to the chemokines produced in the tumor microenvironment, that is, CXCL9 and CXCL10, IFN-γ inducible proteins that promote the infiltration and cytolytic function of CTLs.21 Consistent with IFN-g induction, the mRNA levels of CXCL9 and CXCL10 in tumors derived from mice orally treated with MD-Fraction were significantly increased (4.7- and 3.2-fold, respectively, all p<0.001) compared with those in the PBS-treated mice (Fig. 3e). In addition, a greater infiltration of CD4 and CD8+ cells was found in tumors from the MD-Fraction-administered mice (2.6- and 2.9-fold, p=0.04 and 0.009, respectively) compared with the PBS-treated mice (Fig. 3e). These results indicate that MD-Fraction increases IFN-γ related chemokines in the tumor, resulting in the infiltration of antitumor effector cells into the tumor. To confirm that the impact of T cell immunity on MD-Fraction-induced tumor inhibition, we orally administrated MD-Fraction (20 mg/kg) to Balb/c Nude mice with colon-26 cancer and that did not have T cells. In this mouse model, MD-Fraction did not significantly inhibit tumor growth 18 days after the administration (Fig. 3f). These results suggest that T cell immunity is necessary for the antitumor effect of orally administered MD-Fraction.
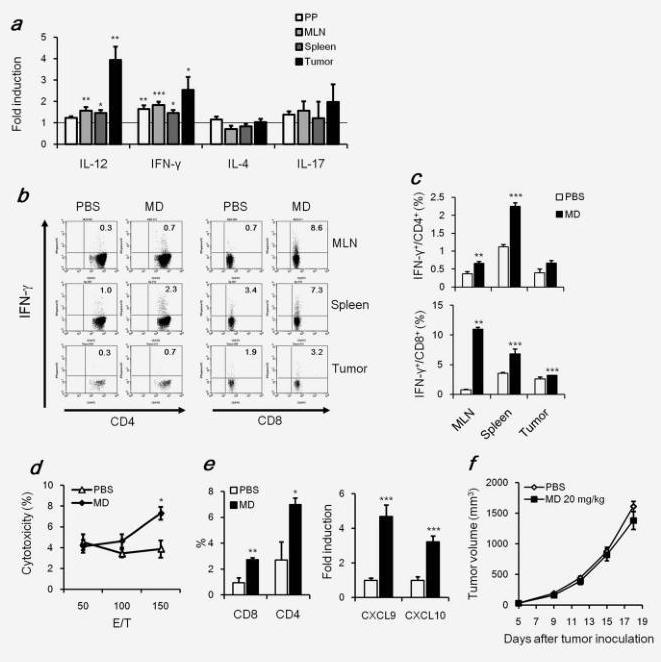
Figure 3. Orally administered MD-Fraction induces the CD4+ and CD8+ T cell response. On day 20 after tumor inoculation, (a) RNA expression levels of Th cytokines in the PP, MLN, spleen and tumor were determined by RT-PCR. The relative expression level in the PP, MLN, spleen and tumor from mice administered MD-Fraction was normalized to the expression level with the PBS control. (b) Flow cytometric analysis showed the expression of IFN-γ in gated CD4+ or CD8+ T cells in the MLN, spleen and tumor. (c) Summarized data of IFN-γ-expressing CD4+ and CD8+ T cells. (d) Cytotoxicity of splenic T cells against colon-26 cells. (e) Expression of CXCL9 and CXCL10 in the tumors was measured by RT-PCR. Relative expression levels of the chemokines were normalized to the expression levels observed for the PBS control mice. Tumor-infiltrating CD4+ and CD8+ cells were analyzed using flow cytometry. (f) Balb/c Nude mice were s.c. Inoculated with colon-26 cells (1 × 105 cells). MD-Fraction was orally administered to the mice 1 day after tumor inoculation, and the tumor growth 0was measured (n=7). The data presented are representative of two independent experiments. *p<0.05, **p<0.01 and ***p<0.001, compared with the PBS control.
MD-Fraction directly induces maturation of DCs via the dectin-1-dependent pathway
Our results showed that orally administered MD-Fraction was captured by macrophages and DCs in the GALT (Fig. 2). Mouse models have demonstrated that the generation of protective anti-tumor immunity depends on the presentation of tumor antigens by DCs.17 To investigate whether MD-Fraction induces antigen presenting function of DCs, we measured the expression of maturation markers involved in Th activation, including CD80, CD86 and MHC class II. As shown in Figure 4a, MD-Fraction significantly induced the expression of surface molecules. Next, we tested whether MD-Fraction-treated DCs produced pro- or anti-inflammatory cytokines required for the Th response. Curdlan, a large particulate β-(1,3)-glucan from Alcaligenes faecalis, induces a robust cytokine response in DCs and is used as a positive control.22 Similar to curdlan, the level of IL-12 p70 production in MD-Fraction-treated DCs (61.6±4.5 pg/mL) was significantly greater than in untreated DCs (10.9±3.1 pg/mL, p<0.001). Interestingly, MD-Fraction did not induce IL-10 expression in the DCs, although curdlan significantly induced IL-10 in DCs (Fig. 4b).
To identify the receptors involved in the DC maturation induced by MD-Fraction, neutralizing antibodies were used. As shown in Figure 4a, the MD-Fraction-induced CD80 and CD86 expression at the DC surface was abrogated by addition of the anti-dectin-1 Ab but not by the anti-CR3 Ab. Consistent with this result, a neutralization assay revealed that MD-Fraction induces IL-12 p70 production in DCs through the dectin-1-dependent pathway (Fig. 4b). TLR2 recognizes zymosan and live fungi and is therefore involved in antifungal immunity.23 We therefore tested the involvement of TLR2 on DC maturation by MD-Fraction. The anti-TLR2 Ab was significantly inhibited IL-12 p70 induced by Pam3CSK4, a typical TLR2 ligand, but did not affect IL-12 p70 induced by MD-Fraction (data not shown). This result indicates that TLR2 is not involved in DC maturation by MD-Fraction.
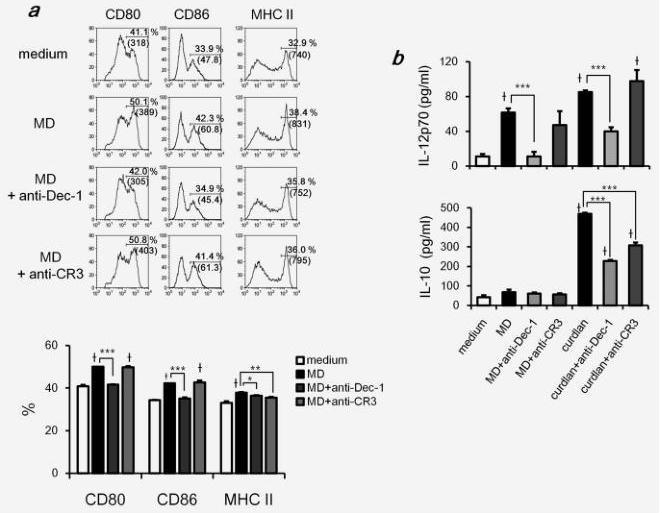
Figure 4. MD-Fraction induces DC maturation. DCs were stimulated with MD-Fraction (400 μg/mL) or curdlan (100 μg/mL) for 24 hr. To block β-glucan receptors, DCs were preincubated with blocking antibodies (5 μg/mL) for 1 hr, followed by stimulation with MD-Fraction or curdlan
for 24 hr. (a) Expression of surface molecules on CD11c+ cells analyzed by flow cytometry. The histograms show the expression of each molecule on CD11c+ DCs. The percentage of positive cells and the fluorescence intensity (MFI) are shown for each histogram. Bar graphs show the percentage of positive cells. (b) IL-12p70 and IL-10 production by MD-Fraction-treated DCs was measured by ELISA. The data presented are representative of three independent experiments. *p<0.05, **p<0.01 and ***p<0.001, compared with the groups linked by the bracket.†p<0.001 compared with untreated DCs.
Orally administered MD-Fraction enhances tumor antigen specific T cell responses against tumor lysate-pulsed DCs
To characterize the influence of the MD-Fraction-induced antitumor activity on the interaction between DCs and T cells, we performed a CFSE dilution assay. Tumor-specific T cells from mice administered MD-Fraction significantly proliferated in response to syngeneic DCs pulsed with colon-26 tumor lysate compared with PBS control mice (Figs. 5a and 5b). In addition, the proliferation of T cells with MD/tumor lysate DCs was higher than the proliferation of T cells with
tumor lysate DCs. Analysis of culture supernatants showed that the IFN-γ production by T cells in MD-Fraction-treated mice was significantly increased in response to MD/lysate-pulsed DCs compared with that in PBS control mice (Fig. 5b). The highest level of IFN-γ was found in T cells in MD-Fraction treated mice with MD/lysate pulsed DCs.
Our results showed that MD-Fraction induces DC maturation through the dectin-1-dependent pathway (Fig. 4). To examine whether MD-Fraction-treated DCs induce the tumor-specific T cell response via dectin-1, we performed a CFSE assay using DCs pretreated with neutralization antibodies. Both proliferation and IFN-γ production by T cells with MD/lysate DCs decreased when blocking with anti-dectin-1 antibody (Figs. 5a and 5b). Notably, dectin-1 inhibition reduced IFN-γ production by T cells with MD/lysate DCs to almost the same level as that observed in T cells with lysate DCs (Fig. 5b). These results indicate that MD-Fraction may induce the tumor-specific T cell response through dectin-1-dependent DC maturation.
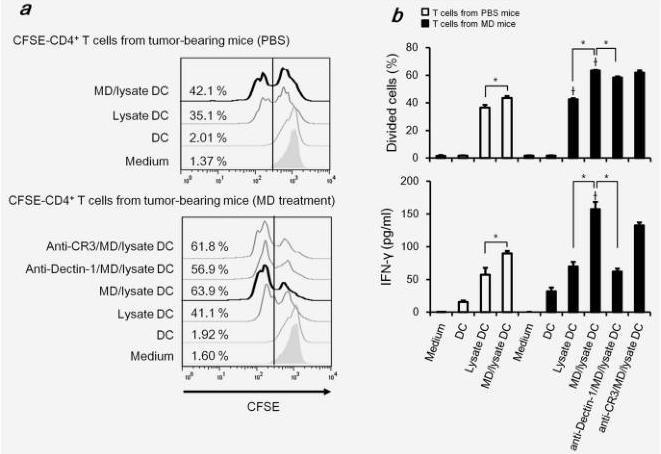
Figure 5. Orally administered MD-Fraction induces the tumor-antigen specific T-cell response. Splenic T cells were isolated from MD-Fraction-administered mice on day 20 after tumor inoculation, and the cells were labeled with CFSE. Syngenic DCs were pulsed with colon-26 lysate and/or MD-Fraction for 24 hr. CFSE-labeled T cells were stimulated with DCs for 72 hr. The fraction of proliferative T cells was measured in a CFSE dilution assay. Representative example (a) and the fraction of divided cells and the concentration of IFN-γ secreted by splenic T cells (b) are shown. The data presented are representative of two independent experiments. *p<0.001 compared with the groups linked by the bracket.†p<0.001 compared with T cells from PBS control mice.
Orally administered MD-Fraction normalizes the tumor microenvironment: effect on Tregs and MDSCs
In advanced cancer, tumor-cell escape can occur through many different mechanisms. One reason of tumor-cell escape is the establishment of an immunosuppressive state within the tumor microenvironment.15 Tregs and MDSCs are two major types of immunosuppressive leukocyte populations that play key roles in inhibiting host-protective antitumor responses. We examined whether orally administered MD-Fraction reduces the immunosuppressive cells. The percentages of Tregs (Foxp3+CD4+) in whole spleen cell suspensions were not altered by MD-Fraction treatment (Fig. 6a). However, MD-Fraction significantly decreased the Tregs in the tumor cell suspension from 0.9 to 0.5% (p=0.001) on day 20 after tumor inoculation (Fig. 6a). In addition, MD-Fraction significantly decreased the ratios Foxp3+CD4+/CD4+ cells in the spleen from 23.3% to 20.8% (p=0.008) and in the tumor from 27.3 to 8.1% (p=0.001) on day 20 after tumor inoculation (Figs. 6a and 6b). These results suggest that MD-Fraction induces effector T cells but not Tregs during tumor growth and that MD-Fraction inhibits the infiltration of Tregs into the tumor sites.
MDSCs are a heterogeneous population commonly expressing both CD11b and Gr-1 markers. MDSCs suppress T and NK cell responses by immune suppressive factors such as arginase 1 (Arg1) and iNOS.16 In tumor-free mice, the percentage of Gr-1+CD11b+ cells in the spleen was slightly increased after oral administration of MD-Fraction (p=0.09; Figs. 6a and 6b). In tumor-bearing mice, although the percentage of Gr-1+CD11b+ cells in the spleen and tumor increased when the tumor growth increased, this percentage of cells was significantly reduced by the oral MD-Fraction treatment in the spleen from 14.2 to 7.9% (p<0.001) and in the tumor from 14.4 to 5.8% (p=0.008) on day 20 after tumor inoculation (Figs. 6a and 6b). Furthermore, we tested the MDSC-related genes Arg1 and iNOS in the tumor sites. As shown in Figure 6c, both Arg1 and iNOS expression were reduced in the oral MD-Fraction-treated tumors. These results suggest that MD-Fraction decreases immunosuppressive MDSCs in the spleen and tumor, which may be able to further enhance the efficacy of MD treatment.
Because of the changes of Tregs and MDSCs, oral administration of MD-Fraction may alter immunosuppressive factors in the tumor microenvironment. As shown in Figure 6c, oral MD-Fraction-treated tumor-bearing mice showed significant reductions in inhibitory cytokine levels (IL-10 and TGF-β) in the tumor.
Discussion
In the current study, we demonstrated that oral administration of MD-Fraction significantly inhibited the tumor growth in mouse models. Orally administered MD-Fraction is taken up by APCs present in the intestinal mucosa, which are then transported to the systemic lymphoid tissues.
In most studies, soluble β-glucans have been administered via i.p. or intravenous injection. However, there is increasing evidence that oral administration of soluble β-glucans also shows immunomodulatory activity.10,24 Rice et al.25 demonstrated that three fluorescently labeled, soluble β-glucans, which vary in molecular weights and structures, are rapidly absorbed from the gastrointestinal tract into the systemic circulation and detected in plasma after oral administration
in rats. Sandvik et al.26 showed that a minute fraction of the orally administered soluble β-glucan translocated to circulation. Conversely, particulate β-glucans are captured predominantly by macrophages or mucosal M cells and are then transported to the bone marrow, spleen and LNs.27,28 After i.p. administration, DTAF-MD-Fraction was immediately transferred into peripheral blood and detected in APCs in peritoneal, spleen, peripheral blood and bone marrow (unpublished observation). In addition, i.p.-administrated MD-Fraction induced IL-12, IFN-γ, GM-CSF and G-CSF in the spleen, resulting the proliferation and activation of systemic immunocompetent cells within 24 hr.7 In contrast, orally administered MD-Fraction was not directly adsorbed and transferred to the blood but was captured by APCs in the GALT, followed by transfer to systemic immune organs starting 3 days after oral administration (Fig. 2a). We speculate that the mucosal APCs the capture MD-Fraction migrate via the afferent lymphatics to the MLNs, where immune modulation is initiated. Twenty days after oral administration of MD-Fraction, the total number of immunocompetent cells remained unchanged. However, the percentage of effector T cells, involved in antitumor immunity, increased. These facts suggest that oral administration of MD-Fraction has a lower risk of excessive immune response than i.p. Administration. However, it is unclear whether MD-Fraction maintains its β-glucan structure after oral administration. More studies are needed to demonstrate whether the β-glucan structure of MD-Fraction is maintained after oral administration, how long the β-glucans remain in the body, and how the β-glucans are cleared.
Particulate β-glucans induce the differentiation of Th1 and Th17 CD4+ T cells via the dectin-1 pathway in DCs.29,30 Soluble PGG β-glucan (MW: 120 kDa) isolated from S. cerevisiae significantly augments the antitumor monoclonal antibody-mediated therapeutic efficacy via CR3.29,32 Thus, the different β-glucans, which vary in origin, molecular structure, solubility and purity, may be recognized by different receptors, resulting in different immune responses.33 Recent studies have shown that soluble b-glucans (MW: ~400 kDa) do not activate dectin-1 signaling.31,34 However, some soluble β-glucans, such as scleroglucan and Sparassis crispa β-glucan (SCG) which have high molecular weights (MW: 1,000 and over 2,000 kDa, respectively), activate bone marrow-derived DCs in a dectin-1-dependent manner.35,36 In this study, we demonstrated that MD-Fraction (MW: 1,200~2,000 kDa) enhances the expression of surface molecules, including CD80, CD86 and MHC class II, as well as secretion of IL-12 p70 by DCs through the dectin-1-dependent pathway (Figs. 4a and 4b). The small, soluble β-glucan (6-8 kDa), laminarin, has been traditionally used as an antagonist of the β-glucan receptor. We confirmed that laminarin did not induce IL-12p70 production in DCs (data not shown). These results suggest that soluble β-glucans of high molecular weights may behave like a particular β-glucans on DCs. We previously demonstrated that MD-Fraction directly induces GM-CSF production by resident macrophages, which can lead to enhanced dectin-1-mediated TNF-α induction and cell proliferation.8 In contrast to the DC, MD-Fraction increased the expression of CD80, CD86 and MHC class II in resident macrophages via the dectin-1-independent pathway (data not shown). In addition, the level of IL-12p70 by resident macrophages was not detectable by stimulation with MD-Fraction (data not shown). Thus, the responses to MD-Fraction may be different between DCs and macrophages. Priming of macrophages with GM-CSF has been reported to induce the dectin-1-mediated inflammatory response.37,38 Because bone marrow derived DCs are differentiated with GM-CSF, induction of dectin-1 signaling by MD-Fraction may occur.
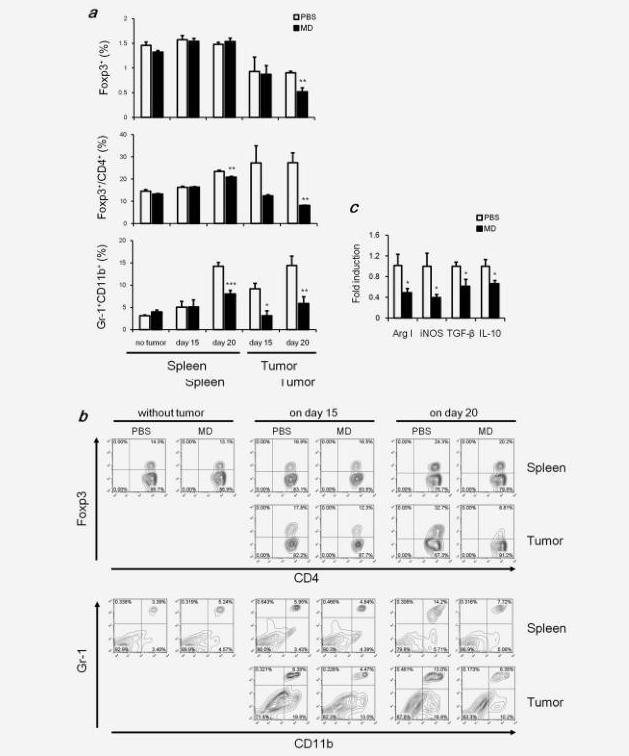
Figure 6. Orally administered MD-Fraction reduces Tregs and MDSCs. Tumor-free mice were orally administered MD-Fraction for 5 days, and splenocytes were harvested. Tumor-bearing mice were orally administered MD-Fraction, and splenocytes and tumor suspensions were harvested on days 15 and 20. The cells were stained using specific antibodies. (a) Summarized data for the percentage of Foxp3-expressing CD4+ cells in a whole-cell suspension or the Foxp3+CD4+/CD4+ ratio, and the percentage of MDSCs and (b) representative image. (c) Tumors on day 20 after tumor inoculation were analyzed for Arg1, iNOS, TGF-β and IL-10 mRNA levels by quantitative RT-PCR. The data (n=5-6) presented are representative of two independent experiments. *p<0.05, **p<0.01, and ***p<0.001, compared with the PBS control.
Successful cancer immunotherapy induces the tumor-specific T cell response to a type 1 cytokine profile.15,39 Orally administered MD-Fraction not only induced IL-12 and IFN-γ expression in the GALT but also induced the expression of these cytokines in the spleen and tumor (Fig. 3). Furthermore, DCs treated with MD-Fraction increased tumor antigen-specific IFN-γ production by splenic T cells in the mice treated with MD-Fraction; this expression was abrogated by the addition of anti-dectin-1 Abs (Fig. 5). In summary, MD-Fraction might induce the tumor antigen-specific T cell response through DC maturation and IL-12 production via the dectin-1-dependent pathway. Generation of effective antitumor immunity during infiltration of effector cells to the tumor mass and execution of effector functions by the antigen-specific T cells to kill the tumor cells are important.40,41 CXCL9 and CXCL10 are chemokines that induce the infiltration of activated T cells to tumors, eventually causing tumor regression.21,42 Tumors from MD-Fraction-administered mice revealed significant increases in CXCL9 and CXCL10 (Fig. 3e). Consistent with the enhancement of expression of these chemokines, orally administered MD-Fraction significantly increased the infiltration of CD8+ T cells to the tumor sites (Fig. 3e). These results suggest that chemokines may recruit CD8+ T cells to the tumor sites to amplify the Th1 antitumor-cellular immunity during oral MD-Fraction therapy.
Immunogenic DCs produce high levels of IL-12, induce the production of IFN-γ and favor the Th1 response that bridges innate and adaptive immunity.17,43 Conversely, following the exposure to IL-10 or TGF-β produced by the tumor, DCs can initiate the development of Tregs that limit these effector responses.17,44,45 In addition, DCs secrete IL-10, which acts in an autocrine manner to suppress the APC functions.46 In this study, we demonstrated that MD-Fraction increased the level of IL-12p70 but not IL-10 by DCs in vitro (Fig. 4b). In addition, the mRNA expression of IL-10 and TGF-β in the tumors decreased significantly after oral administration of MD-Fraction (Fig. 6c). Consistent with these results, the Foxp3+CD4+/CD4+ ratio cells in the spleen and tumor was significantly reduced by MD-Fraction (Figs. 6a and 6b). The reduction of the Treg/CD4 ratio may be partially due to the induction of immunogenic DCs rather than tolerogenic DCs.
MDSCs are characterized by the coexpression of Gr-1 (also known as a granulocyte marker) and CD11b.16 MDSCs were thought to be produced in the bone marrow.47 However, a recent study showed that the spleen is an important extramedullary site that can continuously supply growing tumors with MDSCs.48 Although Gr-1+CD11b+cells in the spleen and tumor from tumor-bearing mice increased when the tumor growth increased, those cells were significantly reduced by the oral MD-Fraction treatment (Fig. 6). The increased number of Gr-1+CD11b+ cells in the spleen on day 20 after tumor inoculation may be immunosuppressive cells induced by the advanced tumor. Our results showed that MD-Fraction decreases immunosuppressive MDSCs in the spleen and tumor, which may be able to further enhance the efficacy of MD treatment. However, how MD-Fraction therapy reduces the MDSC numbers in the tumor-bearing mice remains unknown. Further studies are required to clarify the inhibition mechanism of MDSC by MD-Fraction.
In summary, our results demonstrated that orally administered MD-Fraction inhibits tumor growth by (i) inducing the systemic tumor-antigen specific T cell response via the dectin-1 dependent activation of DCs, (ii) increasing the infiltration of the activated T cells into the tumor and (iii) decreasing tumor-linked immunosuppressive elements such as Tregs and MDSCs. Our preclinical study advocates the implementation of MD-Fraction as an oral therapeutic agent in the management of patients with cancer.
Acknowledgement
The authors thank Yukiguni Maitake Co. for supplying Maitake (G. frondosa).
References
1. Vollmer J, Krieg AM. Immunotherapeutic applications of CpG oligodeoxynucleotide TLR9 agonists. Adv Drug Deliv Rev 2009;61:195-204.
2. Jung ID, Jeong SK, Lee CM, et al. Enhanced efficacy of therapeutic cancer vaccines produced by co-treatment with Mycobacterium tuberculosis heparin-binding hemagglutinin, a novel TLR4 agonist. Cancer Res 2011;71:2858-70.
3. Chihara G, Hamuro J, Maeda Y, et al. Fractionation and purification of the polysaccharides with
marked antitumor activity, especially lentinan, from Lentinus edodes (Berk.) Sing. (an edible mushroom). Cancer Res 1970;30:2776-81.
4. Lu H, Yang Y, Gad E, et al. Polysaccharide krestin is a novel TLR2 agonist that mediates inhibition of tumor growth via stimulation of CD8 T cells and NK cells. Clin Cancer Res 2011;17:67-76.
5. Tsoni SV, Brown GD. beta-Glucans and dectin-1. Ann N Y Acad Sci 2008;1143:45-60.
6. Nanba H, Hamaguchi A, Kuroda H. The chemical structure of an antitumor polysaccharide in fruit bodies of Grifola frondosa (maitake). Chem Pharm Bull (Tokyo) 1987;35:1162-8.
7. Masuda Y, Inoue M, Miyata A, et al. Maitake beta-glucan enhances therapeutic effect and reduces myelosupression and nephrotoxicity of cisplatin in mice. Int Immunopharmacol 2009;9:620-6.
8. Masuda Y, Togo T, Mizuno S, et al. Soluble beta-glucan from Grifola frondosa induces proliferation and Dectin-1/Syk signaling in resident macrophages via the GM-CSF autocrine pathway. J Leukoc Biol 2012;91:547-56.
9. Ito K, Masuda Y, Yamasaki Y, et al. Maitake beta-glucan enhances granulopoiesis and mobilization of granulocytes by increasing G-CSF production and modulating CXCR4/SDF-1 expression. Int Immunopharmacol 2009;9:1189-96.
10. Lin H, de Stanchina E, Zhou XK, et al. Maitake beta-glucan promotes recovery of leukocytes and myeloid cell function in peripheral blood from paclitaxel hematotoxicity. Cancer Immunol Immunother 2010;59:885-97.
11. Deng G, Lin H, Seidman A, et al. A phase I/II trial of a polysaccharide extract from Grifola frondosa (Maitake mushroom) in breast cancer patients: immunological effects. J Cancer Res Clin
Oncol 2009;135:1215-21.
12. Kunisawa J, Kurashima Y, Kiyono H. Gut-associated lymphoid tissues for the development of oral vaccines. Adv Drug Deliv Rev 2011;64:523-30.
13. Grivennikov SI, Greten FR, Karin M. Immunity, inflammation, and cancer. Cell 2010;140:883-99.
14. Rahir G, Moser M. Tumor microenvironment and lymphocyte infiltration. Cancer Immunol
Immunother 2012;61:751-9.
15. Schreiber RD, Old LJ, Smyth MJ. Cancer immunoediting: integrating immunity’s roles in cancer suppression and promotion. Science 2011;331:1565-70.
16. Gabrilovich DI, Nagaraj S. Myeloid-derived suppressor cells as regulators of the immune system. Nat Rev Immunol 2009;9:162-74.
17. Palucka K, Banchereau J. Cancer immunotherapy via dendritic cells. Nat Rev Cancer 2012;12:265-77.
18. Masuda Y, Ito K, Konishi M, et al. A polysaccharide extracted from Grifola frondosa enhances the anti-tumor activity of bone marrow-derived dendritic cell-based immunotherapy against murine colon cancer. Cancer Immunol Immunother 2010;59:1531-41.
19. Qian J, Zheng Y, Zheng C, et al. Active vaccination with Dickkopf-1 induces protective and therapeutic antitumor immunity in murine multiple myeloma. Blood 2012;119:161-9.
20. Masuda Y, Matsumoto A, Toida T, et al. Characterization and antitumor effect of a novel polysaccharide from Grifola frondosa. J Agric Food Chem 2009;57:10143-9.
21. Harlin H, Meng Y, Peterson AC, et al. Chemokine expression in melanoma metastases associated with CD8+ T-cell recruitment. Cancer Res 2009;69:3077-85.
22. Hernanz-Falcon P, Joffre O, Williams DL, Reis e Sousa C. Internalization of Dectin-1 terminates induction of inflammatory responses. Eur J Immunol 2009;39:507-13.
23. Dillon S, Agrawal S, Banerjee K, et al. Yeast zymosan, a stimulus for TLR2 and dectin-1, induces regulatory antigen-presenting cells and immunological tolerance. J Clin Invest 2006;116:916-28.
24. Kamiryo Y, Yajima T, Saito K, et al. Soluble branched (1,4)-beta-D-glucans from Acetobacter
species enhance antitumor activities against MHC class I-negative and -positive malignant melanoma through augmented NK activity and cytotoxic T-cell response. Int J Cancer 2005;115:769-76.
25. Rice PJ, Adams EL, Ozment-Skelton T, et al. Oral delivery and gastrointestinal absorption of soluble glucans stimulate increased resistance to infectious challenge. J Pharmacol Exp Ther 2005;314:1079-86.
26. Sandvik A, Wang YY, Morton HC, et al. Oral and systemic administration of beta-glucan protects against lipopolysaccharide-induced shock and organ injury in rats. Clin Exp Immunol 2007;148:168-77.
27. Hong F, Yan J, Baran JT, et al. Mechanism by which orally administered beta-1,3-glucans enhance the tumoricidal activity of antitumor monoclonal antibodies in murine tumor models. J Immunol 2004;173:797-806.
28. Li B, Cai Y, Qi C, et al. Orally administered particulate beta-glucan modulates tumor-capturing dendritic cells and improves antitumor T-cell responses in cancer. Clin Cancer Res 2010;16:5153-64.
29. Leibundgut-Landmann S, Osorio F, Brown GD, et al. Stimulation of dendritic cells via the dectin-1/Syk pathway allows priming of cytotoxic T-cell responses. Blood 2008;112:4971-80.
30. LeibundGut-Landmann S, Gross O, Robinson MJ, et al. Syk- and CARD9-dependent coupling of innate immunity to the induction of T helper cells that produce interleukin 17. Nat Immunol 2007;8:630-8.
31. Qi C, Cai Y, Gunn L, et al. Differential pathways regulating innate and adaptive antitumor immune responses by particulate and soluble yeast-derived {beta}-glucans. Blood 2011;117:6825-36.
32. Salvador C, Li B, Hansen R, et al. Yeast-derived beta-glucan augments the therapeutic efficacy
mediated by anti-vascular endothelial growth factor monoclonal antibody in human carcinoma xenograft models. Clin Cancer Res 2008;14:1239-47.
33. Palma AS, Feizi T, Zhang Y, et al. Ligands for the beta-glucan receptor, Dectin-1, assigned using "designer" microarrays of oligosaccharide probes (neoglycolipids) generated from glucan polysaccharides. J Biol Chem 2006;281:5771-9.
34. Goodridge HS, Reyes CN, Becker CA, et al. Activation of the innate immune receptor Dectin-1 upon formation of a’phagocytic synapse’. Nature 2011;472:471-5.
35. Huang H, Ostroff GR, Lee CK, et al. Distinct patterns of dendritic cell cytokine release stimulated by fungal beta-glucans and toll-like receptor agonists. Infect Immun 2009;77:1774-81.
36. Harada T, Miura NN, Adachi Y, et al. Highly expressed dectin-1 on bone marrow-derived dendritic cells regulates the sensitivity to beta-glucan in DBA/2 mice. J Interferon Cytokine Res
2008;28:477-86.
37. Goodridge HS, Shimada T, Wolf AJ, et al. Differential use of CARD9 by dectin-1 in macrophages and dendritic cells. J Immunol 2009;182: 1146-54.
38. Parti RP, Loper R, Brown GD, et al. Cytosolic phospholipase a2 activation by Candida albicans in alveolar macrophages: role of dectin-1. Am J Respir Cell Mol Biol 2010;42:415-23.
39. Qu Y, Chen L, Pardee AD, et al. Intralesional delivery of dendritic cells engineered to express
T-bet promotes protective type 1 immunity and the normalization of the tumor microenvironment. J Immunol 2010;185:2895-902.
40. Pardee AD, McCurry D, Alber S, et al. A therapeutic OX40 agonist dynamically alters dendritic, endothelial, and T cell subsets within the established tumor microenvironment. Cancer Res 2010;70:9041-52.
41. Bos R, Sherman LA. CD4+ T-cell help in the tumor milieu is required for recruitment and cytolytic function of CD8+ T lymphocytes. Cancer Res 2010;70:8368-77.
42. Lin YS, Huang LD, Lin CH, et al. In vitro and in vivo anticancer activity of a synthetic glycolipid as Toll-like receptor 4 (TLR4) activator. J Biol Chem 2011;286:43782-92.
43. Giermasz AS, Urban JA, Nakamura Y, et al. Type-1 polarized dendritic cells primed for high IL-12 production show enhanced activity as cancer vaccines. Cancer Immunol Immunother 2009;58:1329-36.
44. Zhou L, Chong MM, Littman DR. Plasticity of CD4+ T cell lineage differentiation. Immunity 2009;30:646-55.
45. Chang LY, Lin YC, Mahalingam J, et al. Tumor-derived chemokine CCL5 enhances TGF-beta-
mediated killing of CD8(+) T cells in colon cancer by T-regulatory cells. Cancer Res 2012;72:1092-102.
46. Couper KN, Blount DG, Riley EM. IL-10: the master regulator of immunity to infection. J Immunol 2008;180:5771-7.
47. Wu L, Yan C, Czader M, et al. Inhibition of PPARgamma in myeloid-lineage cells induces systemic inflammation, immunosuppression, and tumorigenesis. Blood 2012;119: 115-26.
48. Cortez-Retamozo V, Etzrodt M, Newton A, et al. Origins of tumor-associated macrophages and neutrophils. Proc Natl Acad Sci U S A 2012;109:2491-6.
本文部分内容来源网络,如有侵权请联系删除,谢谢。
本文部分内容来源网络,如有侵权请联系删除,谢谢。
编辑:中润世源
